
Naomi Dirckx, PhD
Assistant Professor
- Phone: 314-362-8597
- Fax: BJCIH Bldg. | 11th floor | Office # 11621
- Email: dirckx@nospam.wustl.edu
Welcome to the Dirckx lab!
Our research focusses on the role of citrate and the osteogenic membranous citrate transporter Solute Carrier Family 13 Member 5 (SLC13A5) in bone mineralization and systemic citrate homeostasis.
Osteoblasts are specialized cells derived from mesenchymal stem cells that synthesize and deposit bone matrix. During the differentiation process, osteoblasts synthesize and secrete the type I collagen and non-collagenous matrix proteins that compose the organic fraction of bone. With further differentiation, calcium and phosphate ions are secreted to initiate the calcification process to form the hydroxyapatite nanocrystals shaping the structural integrity of the bone. Converging evidence from both historical and contemporary studies implicate citrate as a structural component of the apatite-collagen nanocomposite. Citrate, a tricarboxylic acid (TCA) cycle intermediate, is used by all aerobic organisms to produce usable chemical energy and is present at strikingly high concentrations in bone (1-5 wt%). In fact, already in 1941 Dickens discovered that over 80% of total body citrate is stored in the skeleton. Independent studies that used high resolution NMR to model the citrate molecule in the apatite crystal suggest that the degree of incorporation of citrate, as well as its spatial orientation in the mineral structure, is critical for maintaining favorable biomechanical properties. Besides generation in the TCA cycle, citrate can also be imported from extracellular space through transmembranous dicarboxylate transporters. The sodium-dependent citrate transporter Solute Carrier Family 13 Member 5 (Slc13a5) is highly expressed in liver and brain where defective citrate transport is associated with protection against diet-induced insulin resistance and childhood epilepsy respectively. Despite the high content of citrate in bone, the role of Slc13a5 in osteoblasts is not well described.
We have recently shown that osteoblasts use a specialized metabolic pathway to regulate citrate uptake from the extracellular milieu as well as endogenous mitochondrial citrate production and deposition of citrate into bone. This occurred through the coordinated functioning of the sodium-coupled membrane citrate transporter SLC13A5 and a zinc transporter protein ZIP1 (encoded by Slc39a1); SLC13A5 mediated citrate entry from blood and its activity exerted homeostatic control of cytoplasmic citrate, which is an allosteric inhibitor of the rate-limiting glycolytic enzyme phosphofructokinase (PFK). Upon transporter blockade, a reduction of SLC13A5-mediated citrate transport caused an initial drop in cytoplasmic citrate levels, reducing the inhibition on glycolysis and thereby triggering endogenous citrate production from glucose within the TCA cycle. Subsequent citrate cataplerosis and secretion were controlled by ZIP1, which mediated zinc-dependent
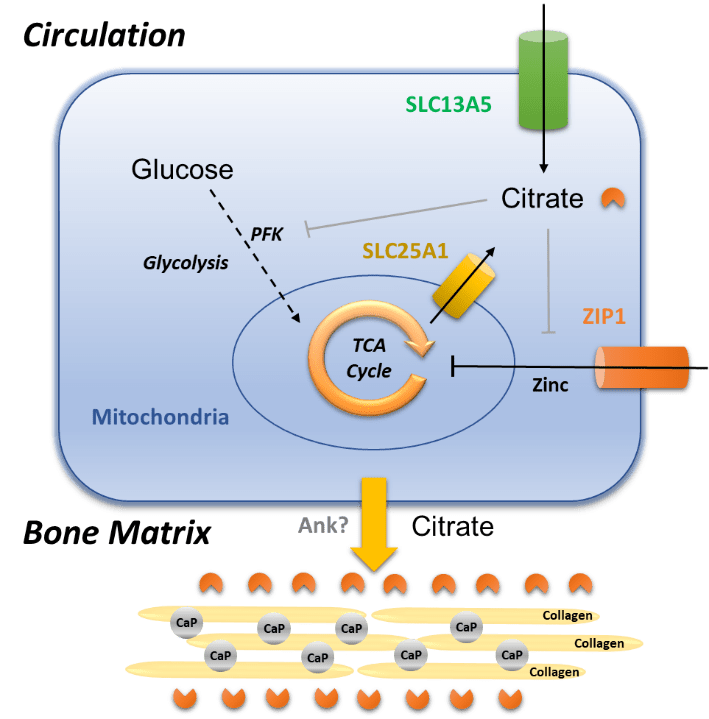
inhibition of the mitochondrial (m-) aconitase enabling the diversion of citrate from the TCA cycle and secretion via the mitochondrial citrate transporter SLC25A1 (See figure).
Loss of SLC13A5 in mice led to elevated systemic citrate levels and a counterintuitive increase in mineral citrate levels both in bones and teeth due to overcompensation of citrate production in and secretion from the mitochondria by upregulation of ZIP1 and SLC25A1. In both global and skeletal-specific knockout mouse models of Slc13a5, increased levels of mineral citrate impaired mineralization, leading to reduced bone mass and strength and hypomineralized teeth, with specifically an absence of enamel. While the latter is a pathological feature that is phenocopied in children with SLC13A5 Loss-Of-Function mutations, it is unknown whether patients with SLC13A5 epilepsy also develop reduced bone volume and strength, and investigation will be challenging due to the secondary effects on bone from weak muscle tone (hypotonia), and lack of motor control (ataxia) in these children. However, by using the combination of global and skeletal-specific knockout mice and in vitro models, it was evident that the origin of the alterations in mineral citrate levels and consequent skeletal hypomineralization was allocated to osteoblasts, rather than other cell types present in the bone or secondary effects from peripheral tissues.
Why is this research important?? Osteoporosis, a low bone mass disorder, is a devastating disease characterized by low bone mineral density and increased fracture risk causing a rising financial and social burden in our aging population. 20% of the elderly population that fracture their hip die within 1 year post fracture and 80% is unable to carry out at least one independent activity of daily living. Therefore it is often called a silent killer as you do not notice your bone loss until you fracture. Within the field we already made significant advances in the generation of pharmaceuticals to improve bone mass in osteoporosis but these treatments do not meet their clinical demands, have their disadvantages and only focus on bone mass while the mechanisms involved in providing favorable mechanical properties to bone remain poorly understood. We believe that understanding the mechanisms bone mineral “citration” could be a first step towards improving bone quality in metabolic bone diseases.
